
Código de barras de ADN - DNA barcoding

El código de barras de ADN es un método de identificación de especies que utiliza una sección corta de ADN de un gen o genes específicos . La premisa de los códigos de barras de ADN es que, en comparación con una biblioteca de referencia de tales secciones de ADN (también llamadas " secuencias "), una secuencia individual puede usarse para identificar de forma única un organismo a una especie, de la misma manera que un escáner de supermercado usa el franjas negras familiares del código de barras UPC para identificar un artículo en su stock con su base de datos de referencia. Estos "códigos de barras" se utilizan a veces en un esfuerzo por identificar especies desconocidas , partes de un organismo o simplemente para catalogar tantos taxones como sea posible, o para comparar con la taxonomía tradicional en un esfuerzo por determinar los límites de las especies.
Se utilizan diferentes regiones de genes para identificar los diferentes grupos de organismos mediante códigos de barras. La región de código de barras más comúnmente utilizada para animales y algunos protistas es una porción del gen de la citocromo c oxidasa I (COI o COX1 ), que se encuentra en el ADN mitocondrial . Otros genes adecuados para el código de barras de ADN son el ARNr del espaciador transcrito interno (ITS) que se usa a menudo para hongos y RuBisCO que se usa para plantas. Los microorganismos se detectan utilizando diferentes regiones de genes. El gen de ARNr 16S , por ejemplo, se usa ampliamente en la identificación de procariotas, mientras que el gen de ARNr 18S se usa principalmente para detectar eucariotas microbianos . Estas regiones de genes se eligen porque tienen menos variación intraespecífica (dentro de la especie) que la variación interespecífica (entre especies), lo que se conoce como "Brecha de código de barras".
Algunas aplicaciones de los códigos de barras de ADN incluyen: identificar las hojas de las plantas incluso cuando las flores o los frutos no están disponibles; identificar el polen recolectado en los cuerpos de los animales polinizadores; identificar larvas de insectos que pueden tener menos caracteres de diagnóstico que los adultos; o investigar la dieta de un animal en función de su contenido estomacal, saliva o heces. Cuando se utilizan códigos de barras para identificar organismos de una muestra que contiene ADN de más de un organismo, se utiliza el término metabarcoding de ADN , por ejemplo, metabarcoding de ADN de comunidades de diatomeas en ríos y arroyos, que se utiliza para evaluar la calidad del agua.
| Parte de una serie sobre |
| Código de barras de ADN |
|---|

|
| Por taxones |
|
| Otro |
Fondo
Las técnicas de códigos de barras de ADN se desarrollaron a partir del trabajo de secuenciación de ADN temprano en comunidades microbianas utilizando el gen de ARNr 5S . En 2003, se propusieron métodos específicos y terminología de códigos de barras de ADN modernos como un método estandarizado para identificar especies, así como para asignar potencialmente secuencias desconocidas a taxones superiores como órdenes y phyla, en un artículo de Paul DN Hebert et al. de la Universidad de Guelph , Ontario , Canadá . Hebert y sus colegas demostraron la utilidad del gen de la citocromo c oxidasa I (COI), utilizado por primera vez por Folmer et al. en 1994, utilizando sus cebadores de ADN publicados como herramienta para análisis filogenéticos a nivel de especie como una herramienta discriminatoria adecuada entre invertebrados metazoarios. La "región de Folmer" del gen COI se usa comúnmente para distinguir entre taxones en función de sus patrones de variación a nivel del ADN. La relativa facilidad para recuperar la secuencia y la variabilidad combinada con la conservación entre especies son algunos de los beneficios de COI. Llamando a los perfiles "códigos de barras", Hebert et al. previó el desarrollo de una base de datos COI que podría servir como base para un "sistema global de bioidentificación".
Metodología
Muestreo y conservación
El código de barras se puede hacer a partir de tejido de una muestra objetivo, de una mezcla de organismos (muestra a granel) o de ADN presente en muestras ambientales (por ejemplo, agua o suelo). Los métodos de muestreo, conservación o análisis difieren entre esos diferentes tipos de muestra.
Muestras de tejido
Para codificar una muestra de tejido de la muestra objetivo, es probable que un pequeño trozo de piel, una escala, una pata o una antena sea suficiente (dependiendo del tamaño de la muestra). Para evitar la contaminación, es necesario esterilizar las herramientas usadas entre muestras. Se recomienda recolectar dos muestras de una muestra, una para archivar y otra para el proceso de codificación de barras. La conservación de la muestra es fundamental para superar el problema de la degradación del ADN.
Muestras a granel
Una muestra a granel es un tipo de muestra ambiental que contiene varios organismos del grupo taxonómico en estudio. La diferencia entre las muestras a granel (en el sentido utilizado aquí) y otras muestras ambientales es que la muestra a granel normalmente proporciona una gran cantidad de ADN de buena calidad. Ejemplos de muestras a granel incluyen muestras de macroinvertebrados acuáticos recolectadas con una red o muestras de insectos recolectadas con una trampa Malaise. Las muestras de agua filtradas o fraccionadas por tamaño que contienen organismos completos como eucariotas unicelulares también se definen a veces como muestras a granel. Estas muestras pueden recolectarse mediante las mismas técnicas que se utilizan para obtener muestras tradicionales para la identificación basada en la morfología.
muestras de eDNA
El método de ADN ambiental (eDNA) es un enfoque no invasivo para detectar e identificar especies a partir de desechos celulares o ADN extracelular presentes en muestras ambientales (por ejemplo, agua o suelo) a través de códigos de barras o metabarcoding. El enfoque se basa en el hecho de que cada organismo vivo deja ADN en el medio ambiente, y este ADN ambiental se puede detectar incluso en organismos que tienen una abundancia muy baja. Por lo tanto, para el muestreo de campo, la parte más crucial es utilizar material y herramientas sin ADN en cada sitio de muestreo o muestra para evitar la contaminación, si es probable que el ADN de los organismos objetivo esté presente en pequeñas cantidades. Por otro lado, una muestra de eDNA siempre incluye el ADN de microorganismos vivos de células completas, que a menudo están presentes en grandes cantidades. Por lo tanto, las muestras de microorganismos tomadas en el entorno natural también se denominan muestras de eDNA, pero la contaminación es menos problemática en este contexto debido a la gran cantidad de organismos objetivo. El método eDNA se aplica a la mayoría de los tipos de muestras, como agua, sedimentos, suelo, heces de animales, contenido estomacal o sangre de, por ejemplo, sanguijuelas.
Extracción, amplificación y secuenciación de ADN
Los códigos de barras de ADN requieren que se extraiga el ADN de la muestra. Existen varios métodos diferentes de extracción de ADN y factores como el costo, el tiempo, el tipo de muestra y el rendimiento afectan la selección del método óptimo.
Cuando el ADN de muestras de organismos o de ADN electrónico se amplifica mediante la reacción en cadena de la polimerasa (PCR), la reacción puede verse afectada negativamente por las moléculas inhibidoras contenidas en la muestra. La eliminación de estos inhibidores es fundamental para garantizar que el ADN de alta calidad esté disponible para análisis posteriores.
La amplificación del ADN extraído es un paso obligatorio en los códigos de barras de ADN. Por lo general, solo se secuencia un pequeño fragmento del material de ADN total (generalmente 400 a 800 pares de bases ) para obtener el código de barras de ADN. La amplificación del material de eDNA generalmente se enfoca en fragmentos de menor tamaño (<200 pares de bases), ya que es más probable que el eDNA esté fragmentado que el material de ADN de otras fuentes. Sin embargo, algunos estudios sostienen que no existe relación entre el tamaño del amplicón y la tasa de detección de eDNA.

Cuando se ha amplificado la región del marcador de código de barras de ADN, el siguiente paso es secuenciar la región del marcador utilizando métodos de secuenciación de ADN . Hay muchas plataformas de secuenciación diferentes disponibles y el desarrollo técnico avanza rápidamente.
Selección de marcador

Los marcadores utilizados para los códigos de barras de ADN se denominan códigos de barras. Para caracterizar con éxito especies basadas en códigos de barras de ADN, la selección de regiones de ADN informativas es crucial. Un buen código de barras de ADN debe tener baja variabilidad intraespecífica y alta interespecífica y poseer sitios flanqueantes conservados para desarrollar cebadores de PCR universales para una amplia aplicación taxonómica . El objetivo es diseñar cebadores que detecten y distingan la mayoría o todas las especies del grupo de organismos estudiado (alta resolución taxonómica). La longitud de la secuencia del código de barras debe ser lo suficientemente corta para ser utilizada con la fuente de muestreo actual, extracción de ADN , amplificación y métodos de secuenciación .
Idealmente, se usaría una secuencia de genes para todos los grupos taxonómicos, desde virus hasta plantas y animales . Sin embargo, aún no se ha encontrado tal región genética, por lo que se utilizan diferentes códigos de barras para diferentes grupos de organismos, o según la pregunta del estudio.
Para los animales , el código de barras más utilizado es el locus de la citocromo C oxidasa I ( COI ) mitocondrial . También se utilizan otros genes mitocondriales, como Cytb , 12S o 18S . Los genes mitocondriales se prefieren a los genes nucleares debido a su falta de intrones , su modo de herencia haploide y su recombinación limitada . Además, cada célula tiene varias mitocondrias (hasta varios miles) y cada una de ellas contiene varias moléculas de ADN circulares . Por lo tanto, las mitocondrias pueden ofrecer una fuente abundante de ADN incluso cuando la muestra de tejido es limitada.
En las plantas , sin embargo, los genes mitocondriales no son apropiados para los códigos de barras del ADN porque exhiben bajas tasas de mutación . Se han encontrado algunos genes candidatos en el genoma del cloroplasto , siendo el más prometedor el gen de la maturasa K ( matK ) solo o en asociación con otros genes. Para la identificación de especies también se han utilizado marcadores de múltiples locus como los espaciadores transcritos internos ribosómicos (ITS DNA) junto con matK , rbcL , trnH u otros genes. La mejor discriminación entre especies de plantas se ha logrado cuando se utilizan dos o más códigos de barras de cloroplasto.
Para las bacterias , la subunidad pequeña del gen del ARN ribosómico ( 16S ) se puede utilizar para diferentes taxones, ya que está muy conservada. Algunos estudios sugieren que COI , chaperonina tipo II ( cpn60 ) o subunidad β de la ARN polimerasa ( rpoB ) también podrían servir como códigos de barras de ADN bacteriano.
Los hongos en los códigos de barras son más desafiantes y es posible que se requiera más de una combinación de cebadores. El marcador COI funciona bien en ciertos grupos de hongos, pero no igualmente bien en otros. Por lo tanto, se están utilizando marcadores adicionales, como ITS rDNA y la gran subunidad de RNA ribosomal nuclear (28S LSU rRNA).
Dentro del grupo de protistas , se han propuesto varios códigos de barras, como las regiones D1-D2 o D2-D3 del rDNA 28S , la subregión V4 del gen rRNA 18S , ITS rDNA y COI . Además, se pueden usar algunos códigos de barras específicos para protistas fotosintéticos , por ejemplo, la subunidad grande del gen de ribulosa-1,5-bisfosfato carboxilasa oxigenasa ( rbcL ) y el gen de ARNr 23S del cloroplástico .
| Grupo de organismos | Gen / locus marcador |
|---|---|
| Animales | COI , Cytb, 12S, 16S |
| Plantas | matK , rbcL , psbA-trnH , su |
| Bacterias | COI , rpoB , 16S, cpn60 , tuf , RIF , tierra |
| Hongos | SU , TEF1α , RPB1 , RPB2 , 18S , 28S |
| Protistas | SU , COI , rbcL , 18S , 28S , 23S |
Bibliotecas de referencia y bioinformática
Las bibliotecas de referencia se utilizan para la identificación taxonómica, también llamada anotación, de secuencias obtenidas de códigos de barras o metabarcos. Estas bases de datos contienen los códigos de barras de ADN asignados a taxones previamente identificados. La mayoría de las bibliotecas de referencia no cubren todas las especies dentro de un grupo de organismos y continuamente se crean nuevas entradas. En el caso de macro y muchos microorganismos (como las algas), estas bibliotecas de referencia requieren documentación detallada (lugar y fecha de muestreo, persona que lo recogió, imagen, etc.) e identificación taxonómica autorizada del espécimen del comprobante, así como la presentación. de secuencias en un formato particular. Sin embargo, estos estándares se cumplen solo para un pequeño número de especies. El proceso también requiere el almacenamiento de especímenes de vales en colecciones de museos, herbarios y otras instituciones colaboradoras. Tanto la cobertura taxonómicamente completa como la calidad del contenido son importantes para la precisión de la identificación. En el mundo microbiano, no hay información de ADN para la mayoría de los nombres de especies, y muchas secuencias de ADN no pueden asignarse a ningún binomio de Linneo . Existen varias bases de datos de referencia según el grupo de organismos y el marcador genético utilizado. Hay bases de datos nacionales más pequeñas (por ejemplo, FinBOL) y grandes consorcios como el Proyecto Internacional de Código de Barras de la Vida (iBOL).
Lanzado en 2007, el Barcode of Life Data System (BOLD) es una de las bases de datos más grandes, que contiene más de 450 000 BIN (Números de índice de código de barras) en 2019. Es un depósito de libre acceso para los registros de muestras y secuencias para estudios de códigos de barras. y también es un banco de trabajo que ayuda a la gestión, aseguramiento de la calidad y análisis de datos de códigos de barras. La base de datos contiene principalmente registros BIN para animales basados en el marcador genético COI.
La base de datos UNITE se lanzó en 2003 y es una base de datos de referencia para la identificación molecular de especies de hongos con la región del marcador genético del espaciador transcrito interno (ITS). Esta base de datos se basa en el concepto de hipótesis de especies: usted elige el% en el que desea trabajar y las secuencias se ordenan en comparación con las secuencias obtenidas a partir de especímenes de comprobantes identificados por expertos.
La base de datos de código de barras Diat.se publicó por primera vez bajo el nombre R-syst :: diatom en 2016 a partir de datos de dos fuentes: la colección de cultivos de Thonon (TCC) en la estación hidrobiológica del Instituto Nacional Francés de Investigación Agrícola (INRA), y de la base de datos de nucleótidos del NCBI (Centro Nacional de Información Biotecnológica). Diat.barcode proporciona datos para dos marcadores genéticos, rbc L (ribulosa-1,5-bisfosfato carboxilasa / oxigenasa) y 18S (ARN ribosómico 18S). La base de datos también incluye información adicional sobre rasgos de las especies, como características morfológicas (biovolumen, dimensiones de tamaño, etc.), formas de vida (movilidad, tipo de colonia, etc.) o características ecológicas (sensibilidad a la contaminación, etc.).
Análisis bioinformático
Para obtener datos bien estructurados, limpios e interpretables, los datos de secuenciación sin procesar deben procesarse mediante análisis bioinformático. El archivo FASTQ con los datos de secuenciación contiene dos tipos de información: las secuencias detectadas en la muestra ( archivo FASTA ) y un archivo de calidad con puntajes de calidad ( puntajes PHRED ) asociados con cada nucleótido de cada secuencia de ADN. Las puntuaciones PHRED indican la probabilidad con la que se ha puntuado correctamente el nucleótido asociado.
| 10 | 90% |
| 20 | 99% |
| 30 | 99,9% |
| 40 | 99,99% |
| 50 | 99,999% |
En general, la puntuación PHRED disminuye hacia el final de cada secuencia de ADN. Por lo tanto, algunas tuberías de bioinformática simplemente cortan el final de las secuencias en un umbral definido.
Algunas tecnologías de secuenciación, como MiSeq, utilizan secuenciación de extremo emparejado durante la cual la secuenciación se realiza desde ambas direcciones para producir una mejor calidad. Las secuencias superpuestas se alinean luego en contigs y se fusionan. Por lo general, se combinan varias muestras en una ejecución y cada muestra se caracteriza por un fragmento de ADN corto, la etiqueta. En un paso de demultiplexación, las secuencias se ordenan utilizando estas etiquetas para reensamblar las muestras separadas. Antes de un análisis posterior, las etiquetas y otros adaptadores se eliminan del fragmento de ADN de la secuencia de código de barras. Durante el recorte, se eliminan las secuencias de mala calidad (puntuaciones PHRED bajas), o las secuencias que son mucho más cortas o más largas que el código de barras de ADN objetivo. El siguiente paso de desreplicación es el proceso en el que todas las secuencias filtradas por calidad se contraen en un conjunto de lecturas únicas (unidades de secuencia individuales ISU) con la información de su abundancia en las muestras. Después de eso, las quimeras (es decir, las secuencias compuestas formadas a partir de piezas de origen mixto) se detectan y eliminan. Finalmente, las secuencias se agrupan en OTU (unidades taxonómicas operativas), utilizando una de las muchas estrategias de agrupación. El software bioinformático más utilizado incluye Mothur, Uparse, Qiime, Galaxy, Obitools, JAMP, Barque y DADA2.
Comparar la abundancia de lecturas, es decir, secuencias, entre diferentes muestras sigue siendo un desafío porque tanto el número total de lecturas en una muestra como la cantidad relativa de lecturas de una especie pueden variar entre muestras, métodos u otras variables. A modo de comparación, se puede reducir el número de lecturas de cada muestra al número mínimo de lecturas de las muestras que se van a comparar, un proceso llamado rarefacción. Otra forma es utilizar la relativa abundancia de lecturas.
Identificación de especies y asignación taxonómica
La asignación taxonómica de las OTU a las especies se logra haciendo coincidir las secuencias con las bibliotecas de referencia. La herramienta de búsqueda de alineación local básica (BLAST) se usa comúnmente para identificar regiones de similitud entre secuencias comparando las lecturas de secuencias de la muestra con secuencias en bases de datos de referencia. Si la base de datos de referencia contiene secuencias de las especies relevantes, entonces las secuencias de muestra se pueden identificar a nivel de especie. Si una secuencia no puede coincidir con una entrada de biblioteca de referencia existente, se pueden utilizar códigos de barras de ADN para crear una nueva entrada.
En algunos casos, debido a lo incompleto de las bases de datos de referencia, la identificación solo se puede lograr en niveles taxonómicos más altos, como la asignación a una familia o clase. En algunos grupos de organismos como las bacterias, la asignación taxonómica a nivel de especie a menudo no es posible. En tales casos, se puede asignar una muestra a una unidad taxonómica operativa (OTU) particular .
Aplicaciones
Las aplicaciones de los códigos de barras de ADN incluyen la identificación de nuevas especies , la evaluación de la inocuidad de los alimentos, la identificación y evaluación de especies crípticas, la detección de especies exóticas, la identificación de especies en peligro y amenazadas , la vinculación de los estadios larvarios y de huevos con las especies adultas, la garantía de los derechos de propiedad intelectual de los recursos biológicos, enmarcar planes de gestión global para las estrategias de conservación y dilucidar los nichos de alimentación. Los marcadores de códigos de barras de ADN se pueden aplicar para abordar cuestiones básicas en sistemática, ecología , biología evolutiva y conservación , incluido el ensamblaje de comunidades, redes de interacción de especies , descubrimiento taxonómico y evaluación de áreas prioritarias para la protección ambiental .
Identificación de especies
Secuencias de ADN cortas específicas o marcadores de una región estandarizada del genoma pueden proporcionar un código de barras de ADN para identificar especies. Los métodos moleculares son especialmente útiles cuando los métodos tradicionales no son aplicables. El código de barras de ADN tiene una gran aplicabilidad en la identificación de larvas para las que generalmente hay pocos caracteres de diagnóstico disponibles y en la asociación de diferentes etapas de la vida (por ejemplo, larva y adulto) en muchos animales. La identificación de las especies incluidas en los apéndices de la Convención sobre el Comercio Internacional de Especies Amenazadas ( CITES ) mediante técnicas de códigos de barras se utiliza en el seguimiento del comercio ilegal.
Detección de especies invasoras
Las especies exóticas se pueden detectar mediante códigos de barras. Los códigos de barras pueden ser adecuados para la detección de especies, por ejemplo, en el control de fronteras, donde a menudo no es posible una identificación morfológica rápida y precisa debido a similitudes entre diferentes especies, falta de características de diagnóstico suficientes y / o falta de experiencia taxonómica. Los códigos de barras y metabarcoding también se pueden utilizar para seleccionar ecosistemas en busca de especies invasoras y para distinguir entre una especie invasora y especies nativas morfológicamente similares.
Delimitación de especies crípticas
Los códigos de barras de ADN permiten la identificación y el reconocimiento de especies crípticas . Sin embargo, los resultados de los análisis de códigos de barras de ADN dependen de la elección de métodos analíticos, por lo que el proceso de delimitación de especies crípticas utilizando códigos de barras de ADN puede ser tan subjetivo como cualquier otra forma de taxonomía . Hebert y col. (2004) concluyó que la mariposa Astraptes fulgerator en el noroeste de Costa Rica en realidad consta de 10 especies diferentes. Estos resultados, sin embargo, fueron posteriormente cuestionados por Brower (2006), quien señaló numerosas fallas graves en el análisis y concluyó que los datos originales no podían respaldar más que la posibilidad de tres a siete taxones crípticos en lugar de diez especies crípticas. Smith y col. (2007) utilizaron códigos de barras de ADN de la citocromo c oxidasa I para la identificación de especies de las 20 morfoespecies de moscas parasitoides Belvosia ( Diptera : Tachinidae ) criadas a partir de orugas ( Lepidoptera ) en el Área de Conservación Guanacaste (ACG), noroeste de Costa Rica. Estos autores descubrieron que los códigos de barras aumentan el recuento de especies a 32, al revelar que cada una de las tres especies de parasitoides , previamente consideradas como generalistas, en realidad son matrices de especies crípticas altamente específicas del huésped. Para 15 morfoespecies de poliquetos dentro del bentos antártico profundo estudiadas mediante códigos de barras de ADN, se encontró diversidad críptica en el 50% de los casos. Además, se detectaron 10 morfoespecies previamente pasadas por alto, lo que aumentó la riqueza total de especies en la muestra en un 233%.
.jpg)
Análisis de la dieta y aplicación de la red alimentaria
El código de barras de ADN y la codificación de metabarras pueden ser útiles en estudios de análisis de dietas, y generalmente se usan si las muestras de presas no se pueden identificar en función de los caracteres morfológicos. Existe una variedad de enfoques de muestreo en el análisis de la dieta: la codificación de metabarras de ADN se puede realizar en el contenido del estómago, las heces, la saliva o el análisis de todo el cuerpo. En muestras fecales o contenidos estomacales altamente digeridos, a menudo no es posible distinguir el tejido de una sola especie y, por lo tanto, se puede aplicar metabarcoding en su lugar. Las heces o la saliva representan métodos de muestreo no invasivos, mientras que el análisis de todo el cuerpo a menudo significa que primero se debe matar al individuo. En el caso de los organismos más pequeños, la secuenciación del contenido del estómago se suele realizar mediante la secuenciación del animal completo.
Códigos de barras para la seguridad alimentaria
Los códigos de barras de ADN representan una herramienta esencial para evaluar la calidad de los productos alimenticios. El objetivo es garantizar la trazabilidad alimentaria, minimizar la piratería alimentaria y valorar la producción agroalimentaria local y típica. Otro propósito es salvaguardar la salud pública; por ejemplo, la codificación metabar ofrece la posibilidad de identificar los meros que causan la intoxicación por pescado con ciguatera de los restos de harina, o de separar los hongos venenosos de los comestibles (Ref).
Biomonitoreo y evaluación ecológica
El código de barras de ADN se puede utilizar para evaluar la presencia de especies en peligro de extinción para los esfuerzos de conservación (Ref), o la presencia de especies indicadoras que reflejan condiciones ecológicas específicas (Ref), por ejemplo, exceso de nutrientes o niveles bajos de oxígeno.
Potenciales y deficiencias
Potenciales
Los métodos tradicionales de bioevaluación están bien establecidos a nivel internacional y sirven bien para el biomonitoreo, como por ejemplo para la bioevaluación acuática dentro de las Directivas de la UE WFD y MSFD . Sin embargo, los códigos de barras de ADN podrían mejorar los métodos tradicionales por las siguientes razones; Los códigos de barras de ADN (i) pueden aumentar la resolución taxonómica y armonizar la identificación de taxones que son difíciles de identificar o que carecen de expertos, (ii) pueden relacionar de manera más precisa / precisa los factores ambientales con taxones específicos (iii) pueden aumentar la comparabilidad entre regiones, (iv) permite la inclusión de etapas tempranas de la vida y especímenes fragmentados, (v) permite la delimitación de especies crípticas / raras (vi) permite el desarrollo de nuevos índices, por ejemplo, especies raras / crípticas que pueden ser sensibles / tolerantes a factores estresantes , (vii) aumenta la cantidad de muestras que se pueden procesar y reduce el tiempo de procesamiento, lo que da como resultado un mayor conocimiento de la ecología de las especies, (viii) es una forma no invasiva de monitoreo cuando se utilizan métodos de eDNA .
Tiempo y costo
El código de barras de ADN es más rápido que los métodos morfológicos tradicionales desde el entrenamiento hasta la asignación taxonómica. Se necesita menos tiempo para adquirir experiencia en métodos de ADN que convertirse en un experto en taxonomía. Además, el flujo de trabajo del código de barras de ADN (es decir, de la muestra al resultado) es generalmente más rápido que el flujo de trabajo morfológico tradicional y permite el procesamiento de más muestras.
Resolución taxonómica
Los códigos de barras de ADN permiten la resolución de taxones desde niveles taxonómicos superiores (por ejemplo, familiares) a niveles taxonómicos inferiores (por ejemplo, especies), que de otro modo serían demasiado difíciles de identificar utilizando métodos morfológicos tradicionales, como por ejemplo la identificación mediante microscopía. Por ejemplo, los Chironomidae (el mosquito que no pica) están ampliamente distribuidos en ecosistemas terrestres y de agua dulce. Su riqueza y abundancia los hacen importantes para los procesos y redes ecológicos, y son uno de los muchos grupos de invertebrados utilizados en el biomonitoreo. Las muestras de invertebrados pueden contener hasta 100 especies de quironómidos que a menudo constituyen hasta el 50% de una muestra. A pesar de esto, generalmente no se identifican por debajo del nivel familiar debido a la experiencia taxonómica y el tiempo requerido. Esto puede resultar en diferentes especies de quironómidos con diferentes preferencias ecológicas agrupadas, dando como resultado una evaluación inexacta de la calidad del agua.
Los códigos de barras de ADN brindan la oportunidad de resolver taxones y relacionar directamente los efectos de los estresores con taxones específicos, como las especies de quironómidos individuales. Por ejemplo, Beermann et al. (2018) Chironomidae con código de barras de ADN para investigar su respuesta a múltiples factores estresantes; flujo reducido, aumento de sedimentos finos y aumento de la salinidad. Después del código de barras, se encontró que la muestra de quironómidos consistía en 183 Unidades Taxonómicas Operativas (OTU), es decir, códigos de barras (secuencias) que a menudo son equivalentes a especies morfológicas. Estas 183 OTU mostraron 15 tipos de respuesta en lugar de los dos tipos de respuesta registrados anteriormente cuando todos los quironómidos se agruparon en el mismo estudio de múltiples factores de estrés. Una tendencia similar se descubrió en un estudio de Macher et al. (2016) que descubrió diversidad críptica dentro de la especie de efímera de Nueva Zelanda Deleatidium sp . Este estudio encontró diferentes patrones de respuesta de 12 OTU moleculares distintas a factores estresantes que pueden cambiar el consenso de que esta efímera es sensible a la contaminación.
Defectos
A pesar de las ventajas que ofrecen los códigos de barras de ADN, también se ha sugerido que el código de barras de ADN se utiliza mejor como complemento de los métodos morfológicos tradicionales. Esta recomendación se basa en múltiples desafíos percibidos.
Parámetros físicos
No es completamente sencillo conectar los códigos de barras de ADN con las preferencias ecológicas del taxón con código de barras en cuestión, como es necesario si el código de barras se va a utilizar para el biomonitoreo. Por ejemplo, la detección de ADN diana en sistemas acuáticos depende de la concentración de moléculas de ADN en un sitio, que a su vez puede verse afectado por muchos factores. La presencia de moléculas de ADN también depende de la dispersión en un sitio, por ejemplo, la dirección o la fuerza de las corrientes. No se sabe realmente cómo se mueve el ADN en arroyos y lagos, lo que dificulta el muestreo. Otro factor podría ser el comportamiento de la especie objetivo, por ejemplo, los peces pueden tener cambios estacionales de movimiento, los cangrejos de río o los mejillones liberarán ADN en grandes cantidades solo en determinados momentos de su vida (muda, desove). En el caso del ADN en el suelo, se sabe aún menos sobre distribución, cantidad o calidad.
La principal limitación del método de códigos de barras es que se basa en bibliotecas de referencia de códigos de barras para la identificación taxonómica de las secuencias. La identificación taxonómica es precisa solo si se dispone de una referencia confiable. Sin embargo, la mayoría de las bases de datos aún están incompletas, especialmente para organismos más pequeños, como hongos, fitoplancton, nematodos, etc. Además, las bases de datos actuales contienen identificaciones erróneas, errores ortográficos y otros errores. Hay un esfuerzo masivo de curación y finalización de las bases de datos para todos los organismos necesarios, lo que implica grandes proyectos de códigos de barras (por ejemplo, el proyecto iBOL para la base de datos de referencia de Barcode of Life Data Systems (BOLD)). Sin embargo, completar y curar es difícil y requiere mucho tiempo. Sin muestras acreditadas, no puede haber certeza sobre si la secuencia utilizada como referencia es correcta.
Las bases de datos de secuencias de ADN como GenBank contienen muchas secuencias que no están vinculadas a muestras acreditadas (por ejemplo, muestras de herbario, líneas celulares cultivadas o, a veces, imágenes). Esto es problemático frente a cuestiones taxonómicas como si varias especies deberían dividirse o combinarse, o si las identificaciones pasadas eran sólidas. La reutilización de secuencias, no vinculadas a especímenes avalados, de organismos inicialmente identificados erróneamente puede respaldar conclusiones incorrectas y debe evitarse. Por lo tanto, la mejor práctica para los códigos de barras de ADN es secuenciar las muestras acreditadas. Sin embargo, para muchos taxones puede resultar difícil obtener especímenes de referencia, por ejemplo, con especímenes difíciles de capturar, los especímenes disponibles están mal conservados o se carece de la experiencia taxonómica adecuada.
Es importante destacar que los códigos de barras de ADN también se pueden usar para crear una taxonomía provisional, en cuyo caso las OTU se pueden usar como sustitutos de los binomios latinos tradicionales, lo que reduce significativamente la dependencia de bases de datos de referencia completamente pobladas.
Sesgo tecnológico
Los códigos de barras de ADN también conllevan un sesgo metodológico, desde el muestreo hasta el análisis de datos bioinformáticos . Además del riesgo de contaminación de la muestra de ADN por inhibidores de la PCR, el sesgo del cebador es una de las principales fuentes de errores en los códigos de barras del ADN. El aislamiento de un marcador de ADN eficiente y el diseño de cebadores es un proceso complejo y se ha realizado un esfuerzo considerable para desarrollar cebadores para códigos de barras de ADN en diferentes grupos taxonómicos. Sin embargo, los cebadores a menudo se unirán preferencialmente a algunas secuencias, lo que conducirá a una eficiencia y especificidad diferencial del cebador y una evaluación de las comunidades no representativas y una inflación de la riqueza. Por lo tanto, la composición de las secuencias de comunidades de la muestra se altera principalmente en el paso de PCR. Además, a menudo se requiere la replicación por PCR, pero conduce a un aumento exponencial del riesgo de contaminación. Varios estudios han destacado la posibilidad de utilizar muestras enriquecidas con mitocondrias o enfoques sin PCR para evitar estos sesgos, pero a día de hoy, la técnica de metabarcoding de ADN todavía se basa en la secuenciación de amplicones. Otros sesgos entran en escena durante la secuenciación y durante el procesamiento bioinformático de las secuencias, como la creación de quimeras.
Falta de estandarización
A pesar de que los códigos de barras de ADN se utilizan y aplican más ampliamente, no existe un acuerdo sobre los métodos para la conservación o extracción de ADN, las opciones de marcadores de ADN y conjuntos de cebadores o los protocolos de PCR. Los parámetros de las tuberías de bioinformática (por ejemplo, agrupación de OTU, algoritmos de asignación taxonómica o umbrales, etc.) están en el origen de un gran debate entre los usuarios de códigos de barras de ADN. Las tecnologías de secuenciación también están evolucionando rápidamente, junto con las herramientas para el análisis de las cantidades masivas de datos de ADN generados, y se necesita con urgencia la estandarización de los métodos para permitir la colaboración y el intercambio de datos a mayor escala espacial y temporal. Esta estandarización de los métodos de códigos de barras a escala europea es parte de los objetivos de la European COST Action DNAqua-net y también es abordada por CEN (el Comité Europeo de Normalización).
Otra crítica del código de barras de ADN es su eficiencia limitada para una discriminación precisa por debajo del nivel de especie (por ejemplo, para distinguir entre variedades), para la detección de híbridos, y que puede verse afectado por las tasas de evolución (se necesita Ref.).
Discrepancias entre la identificación convencional (morfológica) y la basada en códigos de barras
Es importante saber que las listas de taxones derivadas de la identificación convencional (morfológica) no son, y tal vez nunca lo serán, directamente comparables a las listas de taxones derivadas de la identificación basada en códigos de barras debido a varias razones. La causa más importante es probablemente la incompletitud y la falta de precisión de las bases de datos de referencia molecular que impiden una correcta asignación taxonómica de las secuencias de eDNA. El eDNA no encontrará taxones que no estén presentes en las bases de datos de referencia, y las secuencias vinculadas a un nombre incorrecto darán lugar a una identificación incorrecta. Otras causas conocidas son una escala y tamaño de muestreo diferente entre una muestra tradicional y una molecular, el posible análisis de organismos muertos, que puede ocurrir de diferentes maneras para ambos métodos dependiendo del grupo de organismos, y la selección específica de identificación en cualquiera de los métodos, es decir, variando la experiencia taxonómica o la posibilidad de identificar ciertos grupos de organismos, respectivamente, sesgo del cebador que conduce también a un posible análisis sesgado de taxones.
Estimaciones de riqueza / diversidad
El código de barras de ADN puede resultar en una sobreestimación o subestimación de la riqueza y diversidad de especies. Algunos estudios sugieren que los artefactos (identificación de especies que no están presentes en una comunidad) son una causa importante de biodiversidad inflada. El tema más problemático son los taxones representados por un bajo número de lecturas de secuenciación. Estas lecturas generalmente se eliminan durante el proceso de filtrado de datos, ya que diferentes estudios sugieren que la mayoría de estas lecturas de baja frecuencia pueden ser artefactos. Sin embargo, pueden existir taxones raros reales entre estas lecturas de baja abundancia. Las secuencias raras pueden reflejar linajes únicos en comunidades que las convierten en secuencias informativas y valiosas. Por lo tanto, existe una gran necesidad de algoritmos bioinformáticos más robustos que permitan la diferenciación entre lecturas informativas y artefactos. Las bibliotecas de referencia completas también permitirían una mejor prueba de los algoritmos bioinformáticos, al permitir un mejor filtrado de los artefactos (es decir, la eliminación de secuencias que carecen de una contraparte entre las especies existentes) y, por lo tanto, sería posible obtener una asignación de especies más precisa. La diversidad críptica también puede resultar en una biodiversidad inflada, ya que una especie morfológica puede dividirse en muchas secuencias moleculares distintas.
Metabarcoding
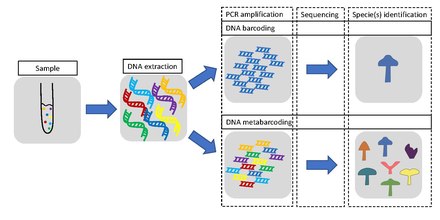
La codificación metabarra se define como el código de barras de ADN o eDNA (ADN ambiental) que permite la identificación simultánea de muchos taxones dentro de la misma muestra (ambiental), aunque a menudo dentro del mismo grupo de organismos. La principal diferencia entre los enfoques es que la codificación de metabarras, a diferencia de los códigos de barras, no se centra en un organismo específico, sino que tiene como objetivo determinar la composición de especies dentro de una muestra.
Metodología
El procedimiento de metabarcoding, como el código de barras general, cubre los pasos de extracción de ADN , amplificación por PCR , secuenciación y análisis de datos . Un código de barras consta de una región de genes variable corta (por ejemplo, ver diferentes marcadores / códigos de barras ) que es útil para la asignación taxonómica flanqueada por regiones de genes altamente conservadas que se pueden utilizar para el diseño de cebadores . Se utilizan diferentes genes dependiendo de si el objetivo es codificar una sola especie con códigos de barras o metabarcodificar varias especies. En el último caso, se utiliza un gen más universal. La metabarcoding no utiliza ADN / ARN de una sola especie como punto de partida, sino ADN / ARN de varios organismos diferentes derivados de una muestra ambiental o global.
Aplicaciones
La metabarcoding tiene el potencial de complementar las medidas de biodiversidad e incluso reemplazarlas en algunos casos, especialmente a medida que la tecnología avanza y los procedimientos se vuelven gradualmente más baratos, optimizados y generalizados.
Las aplicaciones de codificación de metabarras de ADN incluyen:
- Monitoreo de la biodiversidad en ambientes terrestres y acuáticos
- Paleontología y ecosistemas ancestrales
- Interacciones planta-polinizador
- Análisis de dieta
- Seguridad alimenticia
Ventajas y retos
Las ventajas y desventajas generales de los códigos de barras revisadas anteriormente son válidas también para los metabarcoding. Un inconveniente particular de los estudios de metabarcoding es que aún no existe un consenso con respecto al diseño experimental óptimo y los criterios bioinformáticos que se deben aplicar en la metabarcoding de eDNA. Sin embargo, actualmente existen intentos conjuntos, como por ejemplo, la red EU COST DNAqua-Net , para avanzar mediante el intercambio de experiencias y conocimientos para establecer estándares de mejores prácticas para el biomonitoreo.
Ver también
Subtemas:
Temas relacionados:
También vea la barra de navegación lateral en la parte superior del artículo.